Voir la traduction automatique
Ceci est une traduction automatique. Pour voir le texte original en anglais cliquez ici
#Tendances produits
{{{sourceTextContent.title}}}
Quand la partie 11 du titre 21 du CFR est-elle entrée en vigueur ? De quoi s'agit-il et comment s'y conformer ?
{{{sourceTextContent.subTitle}}}
La conformité à la norme 21 CFR Partie 11 est obligatoire pour les secteurs pharmaceutique et des compléments alimentaires réglementés par la FDA. Si vous ne savez pas en quoi elle consiste ni comment vous y conformer, vous trouverez les réponses à vos questions dans cet article.
{{{sourceTextContent.description}}}
Dans les secteurs industriels réglementés, les registres numériques ne sont plus de simples fichiers destinés à stocker des données. Ils sont devenus des sources de preuves traçables.
Pour les entreprises pharmaceutiques, nutraceutiques, de dispositifs médicaux et de fabrication à façon, les registres électroniques et les signatures électroniques sont essentiels pour garantir la sécurité de la production, maintenir l’assurance qualité et prévenir les perturbations coûteuses causées par des modifications de formulation ou des ajustements des paramètres de processus. Au fur et à mesure de l’évolution de la réglementation américaine, la conformité à la norme 21 CFR Part 11 est progressivement devenue une exigence obligatoire plutôt qu’un investissement facultatif.
Pour de nombreux fabricants, en particulier les petites et moyennes entreprises, la mise en œuvre de solutions de validation des systèmes et de gestion des données conformes peut sembler coûteuse et complexe. Par conséquent, deux questions clés sont souvent posées en premier lieu : quand la norme 21 CFR Partie 11 est-elle entrée en vigueur, et qu’est-ce que la conformité à la norme 21 CFR Partie 11 ?
Ce guide complet explique en détail la norme 21 CFR Partie 11. Nous y aborderons ce qu’est la norme 21 CFR Partie 11, qui doit s’y conformer, ses principales exigences, ainsi que la manière d’atteindre la conformité à la norme 21 CFR Partie 11 de manière pratique et rentable.
Points clés à retenir
Date d’entrée en vigueur de la norme 21 CFR Partie 11 et contexte historique.
Qu’est-ce que la norme 21 CFR Partie 11 dans l’industrie pharmaceutique ?
Qui doit se conformer à la norme 21 CFR Partie 11 dans divers secteurs d’activité ?
Exigences fondamentales de la partie 11 du 21 CFR concernant les enregistrements, les signatures, les accès et les pistes d’audit.
Comment se conformer à la partie 11 du 21 CFR, étape par étape.
Comment les équipements conformes à la partie 11 facilitent la production pharmaceutique et nutraceutique.
1. Quand la norme 21 CFR Partie 11 est-elle entrée en vigueur ?
Selon le site officiel de la FDA, la norme 21 CFR Partie 11 est entrée en vigueur le 20 août 1997.
Qu’est-ce qui a motivé la publication de cette réglementation ? Son origine réside dans la transition majeure des registres papier vers les systèmes informatisés. Auparavant, les registres de production pharmaceutique, les rapports de laboratoire et les documents qualité étaient en grande partie créés, signés et stockés sur papier. À mesure que l’échelle de production s’est développée, les simples registres papier ne pouvaient plus répondre au besoin de documentation rapide et de récupération immédiate des données, ce qui a conduit les entreprises à adopter des systèmes numériques.
Afin de garantir la fiabilité de ces nouveaux systèmes, la partie 11 du titre 21 du Code des règlements fédéraux soumet les enregistrements électroniques et les signatures électroniques visés par le 21 CFR 11 à une surveillance réglementaire stricte. Lorsque les entreprises utilisent des données numériques pour étayer des décisions soumises à la réglementation de la FDA, ces données doivent être exactes et sécurisées, mais aussi traçables et accessibles lors des inspections de la FDA.
2. Qu’est-ce que la partie 11 du 21 CFR ?
Définition de la partie 11 du 21 CFR
Fondamentalement, qu’est-ce que la partie 11 du 21 CFR ? Il s’agit d’une réglementation établie par la FDA américaine qui dicte la manière de gérer les enregistrements électroniques et les signatures.
Pour comprendre la réglementation 21 CFR Partie 11, il faut se rappeler que, par le passé, la FDA surveillait les armoires de classement verrouillées, contrôlait qui les gérait et suivait qui en ouvrait ou en modifiait le contenu. Ces armoires de classement ayant évolué vers des systèmes numériques, la FDA a mis en place ces directives de la norme 21 CFR Partie 11 afin de garantir que les systèmes soient correctement contrôlés et que les données restent sécurisées.
Quels types de dossiers sont concernés ?
Les dossiers électroniques relevant de la norme 21 CFR Partie 11 peuvent inclure :
Les dossiers de lots électroniques
Les dossiers de fabrication
Les dossiers de contrôle qualité
Les journaux d’exploitation des équipements
Les résultats d’essais en laboratoire
Les registres de nettoyage et d’entretien
Les registres de formation
Les registres de contrôle des changements et des écarts
Il convient de noter que cette réglementation ne s’applique pas à tous les fichiers numériques ; elle ne s’applique que lorsque des registres électroniques sont utilisés pour satisfaire aux exigences de la FDA en matière de tenue des registres.
Quels secteurs d’activité doivent s’y conformer ?
Les exigences de conformité prévues par le 21 CFR Partie 11 peuvent s’appliquer à un large éventail d’entités, notamment les fabricants de produits pharmaceutiques, de compléments alimentaires et de dispositifs médicaux, ainsi qu’aux organismes de fabrication sous contrat, aux laboratoires d’essais et aux entreprises exportant vers les États-Unis des produits réglementés par la FDA.
Par conséquent, des questions telles que « La norme 21 CFR Partie 11 s’applique-t-elle aux dispositifs médicaux ? » et « Que couvre la norme 21 CFR Partie 11 ? » sont essentielles ; le principe fondamental régissant l’applicabilité de la règle est que les exigences de conformité s’appliquent dès lors que des systèmes électroniques sont utilisés pour prendre en charge des enregistrements ou des autorisations soumis aux exigences de la FDA en matière d’intégrité des données.
3. Pourquoi la norme 21 CFR Partie 11 a-t-elle été introduite ?
La norme 21 CFR Partie 11 a été introduite car la fabrication numérique a engendré à la fois de nouveaux gains d’efficacité et de nouveaux risques en matière de conformité.
La partie 11 du titre 21 du CFR a vu le jour en réponse aux gains d’efficacité et aux nouveaux risques de conformité associés à la fabrication numérique. Alors que le secteur passait des dossiers papier aux systèmes numériques, la FDA a dû établir une réglementation garantissant la fiabilité des dossiers électroniques ; cette règle s’applique aux dossiers réglementés par la FDA qui sont créés, modifiés, conservés, archivés, consultés ou transmis par voie électronique.
L’objectif principal de cette réglementation est de préserver l’intégrité des données relevant de la partie 11 du 21 CFR et d’empêcher toute altération ou falsification en exigeant que les systèmes intègrent des pistes d’audit, fonctionnant à la manière d’une « boîte noire » qui enregistre la séquence exacte des événements. Lors des inspections de la FDA, les autorités de contrôle examinent les dossiers de lots électroniques, les exigences relatives aux pistes d’audit de la partie 11, les contrôles d’accès des utilisateurs et les signatures électroniques conformes à la norme 21 CFR Partie 11 ; ces archives historiques exhaustives constituent la pierre angulaire de la conformité réglementaire, de la qualité des produits et de la confiance des clients.
4. Qui doit se conformer à la norme 21 CFR Partie 11 ?
Toutes les entreprises n’ont pas besoin du même niveau de contrôle, mais de nombreux fabricants soumis à la réglementation de la FDA devraient évaluer qui doit se conformer à la partie 11 du 21 CFR en fonction de leurs systèmes et de leur marché.
Fabricants de médicaments
Dans le secteur pharmaceutique soumis à la partie 11 du 21 CFR, les systèmes d’enregistrements électroniques impliquant des informations sur les lots, le contrôle qualité, les registres d’équipements ou les autorisations de mise sur le marché sont généralement tenus de se conformer à la partie 11 du 21 CFR. Cependant, dès lors que les enregistrements électroniques remplacent officiellement les enregistrements papier, cette obligation de conformité devient impérative et non plus facultative.
Fabricants de compléments alimentaires
Si un fabricant de compléments alimentaires traite électroniquement ses registres de production, de qualité ou de lots et que le produit est destiné à la vente aux États-Unis, la conformité à la norme CFR Partie 11 est susceptible de s’appliquer.
Fabricants de dispositifs médicaux
Pour ceux qui se demandent si la norme 21 CFR Partie 11 s’applique aux dispositifs médicaux, la réponse est oui lorsque les enregistrements électroniques répondent aux exigences du système qualité de la FDA.
Organismes de fabrication sous contrat (CMO)
Les CMO doivent souvent assurer une conformité rigoureuse à la norme 21 CFR Partie 11 de la FDA, car ils travaillent pour plusieurs clients et font l’objet d’audits fréquents de la part de ces derniers.
Entreprises exportant vers les États-Unis
Même si le fabricant est situé en dehors des États-Unis, la conformité aux exigences de la partie 11 reste généralement obligatoire à condition que les produits soient vendus aux États-Unis et que les enregistrements électroniques concernent des activités réglementées par la FDA.
5. Quelles sont les exigences de la partie 11 du 21 CFR ?
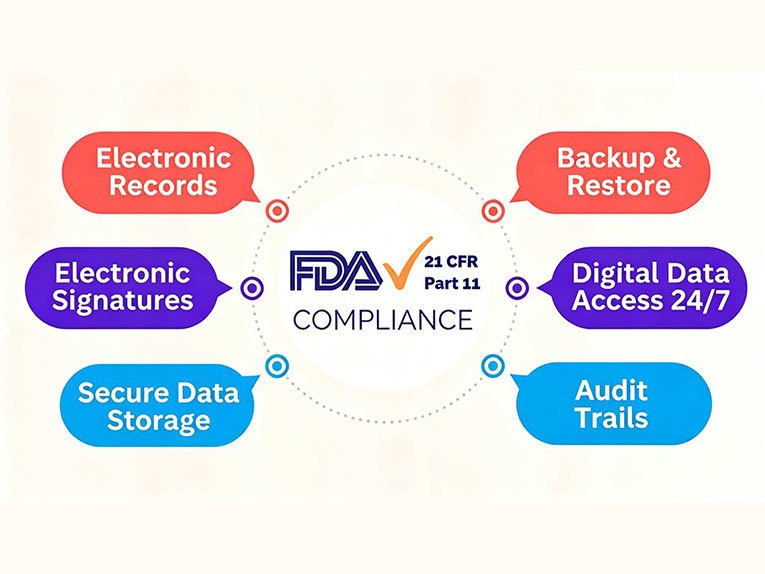
Conformément à ce document, quelles sont exactement les exigences de la partie 11 du titre 21 du CFR ? La réponse peut être regroupée en plusieurs contrôles pratiques. Les exigences de conformité prévues par la partie 11 du titre 21 du CFR s’articulent autour de cinq domaines clés : les enregistrements électroniques, les signatures électroniques, les pistes d’audit, les contrôles d’accès des utilisateurs, ainsi que la conservation et la récupération des enregistrements.
Dossiers électroniques
Les exigences de la norme 21 CFR Partie 11 relatives aux dossiers électroniques stipulent que ceux-ci doivent être exacts, complets, sécurisés, lisibles et récupérables. Les entreprises doivent assurer une conservation adéquate des dossiers et un stockage sécurisé des données.
Signatures électroniques
Conformément à la partie 11 de la réglementation de la FDA, les signatures électroniques doivent clairement établir l’identité et la responsabilité du signataire, en affichant son nom, l’horodatage et la nature de l’action (par exemple, « autorisation de mise sur le marché ») afin de garantir qu’elles aient la même validité juridique que les signatures manuscrites.
Pistes d'audit
Le fonctionnement des pistes d'audit dans le domaine des équipements pharmaceutiques est une question courante. Les pistes d'audit enregistrent automatiquement toutes les actions critiques, notamment la création, la modification, la suppression, l'approbation et les changements apportés au système, afin de garantir la traçabilité. Si les données de poids d’un lot passent de 500 kg à 510 kg, le journal d’audit enregistre l’auteur de la modification, l’heure de la modification ainsi que les valeurs avant et après la modification, ce qui facilite l’analyse des causes profondes.
Contrôles d’accès des utilisateurs
Les contrôles d’accès garantissent que seul le personnel autorisé peut effectuer des actions spécifiques grâce à une protection par mot de passe et à la gestion des autorisations, en recourant à l’authentification multifactorielle et au principe du privilège minimal.
Conservation et récupération des enregistrements
Les enregistrements doivent rester facilement accessibles pendant toute la durée de conservation imposée par la FDA ; l’incapacité à récupérer rapidement les données lors d’un audit est considérée comme une grave violation de la conformité.
6. Comment se conformer à la norme 21 CFR Partie 11
La mise en conformité est une préoccupation majeure pour la plupart des entreprises pharmaceutiques et de produits de santé. Dans cette section, nous allons explorer le sujet en profondeur, en abordant les étapes spécifiques de mise en œuvre, les problèmes potentiels et les solutions correspondantes.
Étape 1 – Réaliser une évaluation de conformité à la norme 21 CFR Partie 11
Vous devez commencer par comparer tous les systèmes qui créent, modifient, stockent, récupèrent ou transmettent des enregistrements électroniques réglementés par la FDA aux directives de la norme 21 CFR Partie 11 applicables aux produits pharmaceutiques. Cette évaluation doit porter sur la validation, la piste d’audit, le contrôle d’accès des utilisateurs, les signatures électroniques, la conservation des enregistrements et la sécurité.
Suivre la liste de contrôle de conformité au 21 CFR Partie 11 peut accélérer votre processus.
Étape 2 – Valider les systèmes informatisés
Un système conforme à la norme 21 CFR Partie 11 doit faire l’objet d’une validation afin de prouver qu’il fonctionne comme prévu. Dans la pratique, la validation doit être menée selon une séquence structurée d’actions :
1. Définir et évaluer la portée et les risques du système
Commencez par documenter clairement ce que le système est censé faire et identifiez où se situent les risques. Par exemple, une machine de remplissage de gélules doit enregistrer le poids de remplissage, la vitesse et le numéro de lot.
2. Tester et vérifier les performances du système
Ensuite, effectuez des tests structurés à l’aide des procédures IQ, OQ et PQ. Cette évaluation vise à vérifier si le système fonctionne comme prévu.
3. Documenter, approuver et maintenir le contrôle
Enfin, toutes les activités de validation doivent être documentées, revues et approuvées avant la mise en service du système.
Étape 3 – Mise en œuvre de la fonctionnalité de piste d’audit
Vous devez garantir l’intégrité de la fonction de piste d’audit, en vous assurant que le système stocke et enregistre de manière sécurisée les données, telles que les horodatages et les modifications, et qu’il permet leur récupération précise. Prenons l’exemple du réglage des paramètres d’une machine de remplissage de gélules : dès qu’un opérateur modifie les paramètres de remplissage, le système enregistre automatiquement les anciennes et les nouvelles valeurs, l’identifiant de l’opérateur et l’horodatage.
Étape 4 – Mettre en place des contrôles relatifs aux signatures électroniques
Pour répondre aux exigences de la FDA en matière de signatures électroniques, chaque utilisateur doit disposer d’une identité unique. Les signatures électroniques doivent être associées à des enregistrements spécifiques et ne doivent pas être copiées ni réattribuées.
Étape 5 – Contrôler l’accès des utilisateurs
Utilisez un accès basé sur les rôles. À chaque rôle doivent correspondre des autorisations différentes. Les administrateurs disposent toujours des autorisations les plus élevées, tandis que les opérateurs ne disposent que d’autorisations partielles.
7. Ruida Packing Machinery : fournisseur de machines conformes aux normes de la FDA et aux BPF
Il arrive parfois que, même si les fabricants souhaitent équiper leurs machines d’un système de piste d’audit, toutes les machines ne le prennent pas en charge. Ruida Packing, en collaboration avec des sociétés pharmaceutiques de renom, notamment UCB et US Pharma, s’engage sans réserve à développer des équipements conformes à la partie 11. Nos machines sont certifiées CE, cGMP, ISO et autres normes associées. En fonction des besoins des clients, Ruida peut également fournir une solution de surveillance conforme à la norme 21 CFR Partie 11 pour les équipements de production et de conditionnement.
Ruida accompagne également la fabrication pharmaceutique grâce à des services concrets :
Matériaux certifiés : acier inoxydable 304 pour les pièces sans contact et acier inoxydable 316L pour les pièces en contact.
Services de mise en service à distance et sur site, avec des centres de service après-vente en Amérique du Nord et à Hong Kong.
Documentation complète sur les machines : manuels, guides d’utilisation, documents de maintenance et vidéos d’assistance après-vente.
Documentation IQ, OQ, PQ : assistance en option pour les documents de validation (IQ, OQ, PQ) et la documentation de conformité.
Pour les fabricants qui prévoient de se mettre en conformité avec la norme 21 CFR Part 11, le choix d’équipements dotés d’options prêtes à la mise en conformité peut réduire les coûts de modification futurs et faciliter la préparation aux audits.
8. Conclusion
En revenant sur la date d’entrée en vigueur de la norme 21 CFR Part 11, on constate à quel point elle a profondément façonné le secteur depuis 1997. Depuis le 20 août de cette année-là, cette réglementation constitue la norme selon laquelle les entreprises soumises à la FDA doivent gérer leurs données électroniques. Aujourd’hui, la conformité à la norme 21 CFR Partie 11 est au cœur des préoccupations de nombreux fabricants de produits pharmaceutiques et de compléments alimentaires, et vous avez tout intérêt à vous y préparer. Non seulement en matière de validation, mais aussi au niveau des machines.
9. FAQ
Q1 : La norme 21 CFR Partie 11 est-elle obligatoire ?
Oui, bien sûr. Mais elle ne s’applique pas à tous les systèmes d’enregistrement électronique. Elle est obligatoire lorsque des enregistrements électroniques ou des signatures électroniques sont utilisés pour répondre aux exigences de la FDA.
Q2 : En quoi les BPF (bonnes pratiques de fabrication) et la norme 21 CFR Partie 11 diffèrent-elles ?
Eh bien, leurs axes prioritaires sont différents. Les BPF mettent l’accent sur la qualité des produits, tandis que la norme 21 CFR Partie 11 porte sur les enregistrements électroniques et les signatures électroniques.
Q3 : Toutes les machines pharmaceutiques doivent-elles être conformes à la Partie 11 ?
Non. Une machine doit être prise en compte au regard de la Partie 11 lorsqu’elle crée, stocke, modifie ou transmet des enregistrements électroniques réglementés. Les équipements purement mécaniques ne nécessitent pas forcément le même niveau de contrôle.
Q4 : Qu’est-ce qu’une piste d’audit ?
Une piste d’audit consiste à enregistrer toutes les actions effectuées sur le système, en indiquant l’action exacte, l’heure et les modifications éventuelles.
Q5 : Comment savoir si un équipement est conforme aux exigences de la Partie 11 ?
Vous pouvez vérifier s’il prend en charge les pistes d’audit, le contrôle d’accès des utilisateurs, les signatures électroniques, le stockage sécurisé des données, l’exportation des enregistrements et les documents de validation. Mais pour faire simple, il suffit de se renseigner auprès des fournisseurs de machines avant l’achat.
[1] Food and Drug Administration (FDA) des États-Unis : Partie 11, Enregistrements électroniques ; Signatures électroniques — Champ d’application et mise en œuvre
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
[2] Code électronique des règlements fédéraux : 21 CFR Partie 11 — Dossiers électroniques ; Signatures électroniques
[3] Journal officiel fédéral : Dossiers électroniques ; Règle définitive sur les signatures électroniques, publiée le 20 mars 1997